A High-Throughput Computational Approach for Catalytic Polyolefin Transformation
Plastics are ubiquitous in modern life and the development of new methods for their recycling and reuse are of paramount importance. Chemical conversion of polyolefins has become an area of extreme interest because of the higher selectivity and retention of mechanical properties of the resulting products compared to other recycling strategies like pyrolysis. Selective and efficient depolymerisation of polyolefins is one of the biggest challenges in the chemical recycling of polymers.
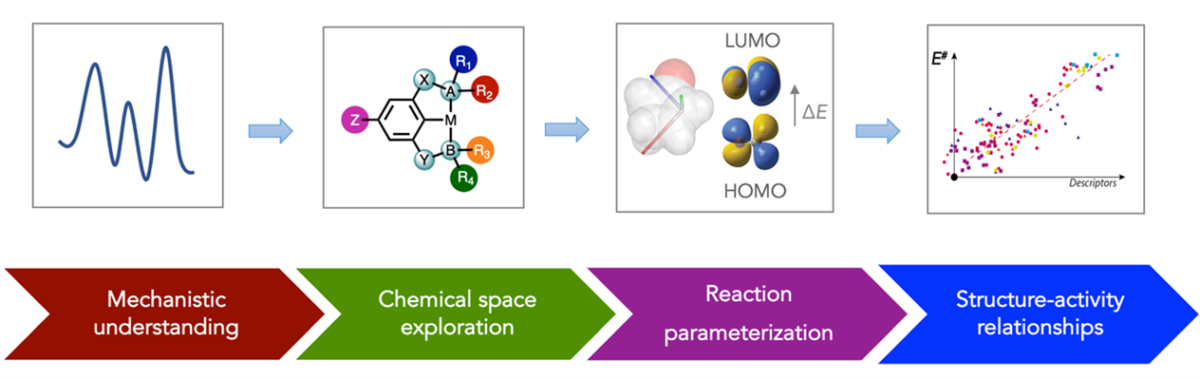
We envisage to develop an automated high-throughput computational approach to address the complex catalytic chemistry of polyolefin transformations in solution or at a solid-liquid interface. Reaction mechanisms of metal catalyzed C-H activation of polyolefin model structures will be computationally studied by using state-of-the-art automated reaction network analysis methods allowing for efficient exploration of various reaction channels accessible in such multicomponent systems. Mechanistic insights will enable the efficient chemical space exploration and calculation of relevant molecular descriptors. The resulting datasets featuring quantum chemical descriptors and structural data for a range of model catalysts, substrates and products combined with computed kinetic and thermodynamic activity metrics will be combined in a regression model to establish a multidimensional universal property-activity relationships for metal-based C-H oxidation catalysts. We anticipate that such an analysis will not only point to key reactivity descriptors to guide the development of superior catalysts, but will also provide information on the effect of substrate functionalization for secondary unselective conversion processes. The computational predictions will be tested and validated in the partner experimental projects within the consortium.


Partners

Share this page